Venturing into new possibilities
Venturing into new possibilities Mines has long been renowned for being innovative in the...


Venturing into new possibilities Mines has long been renowned for being innovative in the...






The best job in the world Some have said that being a university president is the toughest or even...






Supporting new ventures Zack Bennett ’99 has always been an explorer, curious about the unknown...





Supporting new ventures Zack Bennett ’99 has always been an explorer, curious about the unknown...

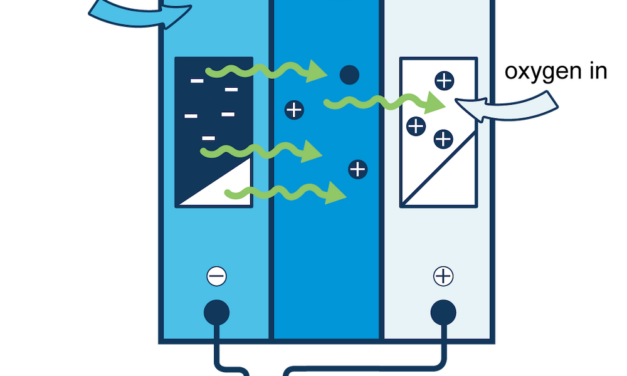
Combining scientific expertise Mines and the U.S. Geological Survey established a joint industry...




Engineering campus When Conor Lenon ’14, MS ’15 was a Mines student, he remembers that the Mines...



The best job in the world Some have said that being a university president is the toughest or even...
Read More
Sweeping success For the first time in program history, Mines’ men’s and women’s track & field...
Read More
In full swing In 1980, the average driving distance of professional golfers on the PGA Tour was...
Read More
Combining scientific expertise Mines and the U.S. Geological Survey established a joint industry...
Read More
Moving the needle for movement sciences There’s still a lot to learn in the field of movement...
Read More
Better care for wound healing Colorado-based biomaterials company GelSana Therapeutics has...
Read More
Supporting new ventures Zack Bennett ’99 has always been an explorer, curious about the unknown...
Read More
Building on traditions, connections and impact The M Climb. Oredigger Camp. Senior Capstone Design...
Read More
Creating a sense of communal support The theme of this year’s Forces of Mines: Elevating Women...
Read More
All in the family As current Mines student Rowan Welch makes his way around the Mines campus, he...
Read More
Leading with integrity Chris Valdez ’00 and Ty Harrison ’98 first met as Mines students and both...
Read More
Engineering campus When Conor Lenon ’14, MS ’15 was a Mines student, he remembers that the Mines...
Read More
Venturing into new possibilities Mines has long been renowned for being innovative in the...
Read More
